
3.1 Sequenciamento metagenômico da região 16S
As comunidades bacterianas são componentes cruciais dos ambientes do solo, com capacidade de degradar produtos químicos tóxicos, como hidrocarbonetos de petróleo (FERNÁNDEZ-LUQUEÑO et al., 2011). Além da diversidade taxonômica, é essencial compreender o potencial funcional da população microbiana presente em áreas contaminadas. Esse conhecimento é importante não só para avaliar se o ambiente pode ser recuperado por processos de atenuação natural, mas também para identificar possíveis vias de biodegradação que possam ser estimuladas. Dessa forma, conhecer o potencial das comunidades microbianas estabelecidas em áreas contaminadas, por meio da inferência funcional, é uma estratégia interessante para prever ou avaliar a aplicabilidade e otimizar os processos de biorremediação.
Nesse aspecto, o sequenciamento da região 16S, utilizando técnicas de segunda geração, permite desvendar uma série de informações importantes sobre a comunidade bacteriana presente em amostras ambientais. Isso possibilita inferir o papel dessas comunidades em diversos processos metabólicos que ocorrem na área de interesse. Em amostras de solo coletadas nas áreas-fonte avaliadas, foram encontrados 38 filos, 103 classes, 211 ordens, 322 famílias, 531 gêneros e 3167 ASV (Amplicon Sequence Variants – Variantes de Sequência de Amplicon) bacterianos, com as mais diversas funcionalidades.
Entre o predomínio de bactérias em nível de Família, foram encontradas Fusobacteriaceae na Área 7 (contaminada com gasolina e 10% de etanol) no monitoramento de fevereiro de 2019 e Oxalobacteraceae em misturas de diesel com etanol, gasolina com etanol e diesel com biodiesel, no monitoramento de janeiro de 2022. A abundância relativa ultrapassou os 50% de Micrococcaceae na Área 10 com diesel e biodiesel. O predomínio de Thermacetogeniaceae foi evidente na Área 6 com 100% de biodiesel e remediada pelo processo de atenuação natural.
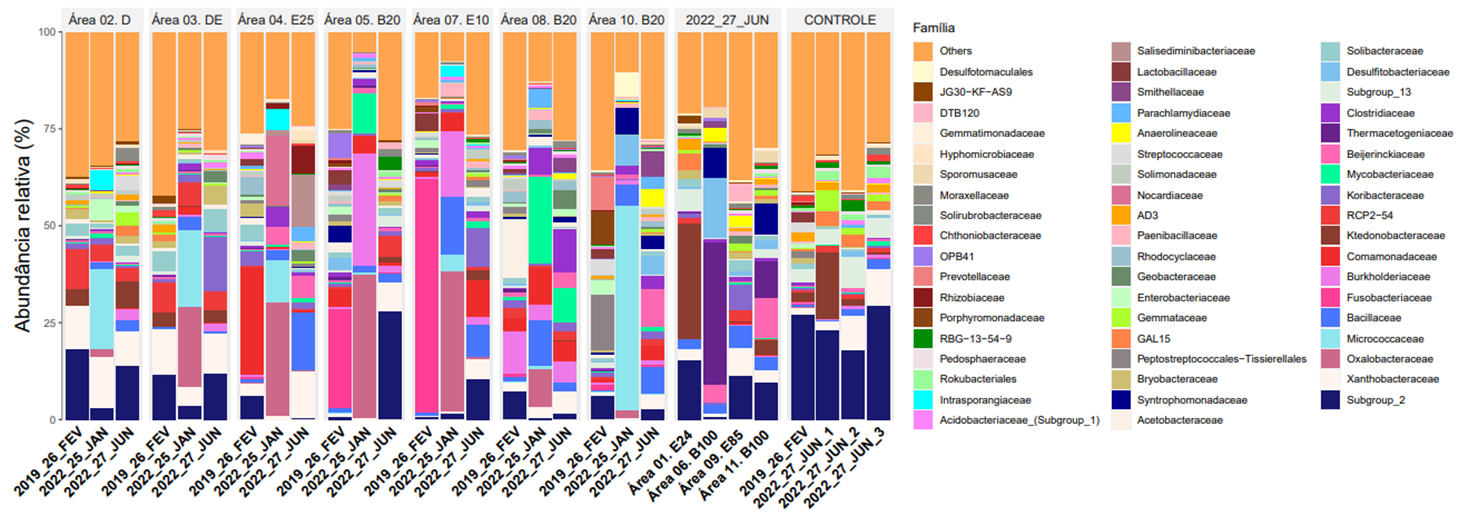
Abundância relativa de bactérias em nível de Família no solo próximo a fonte (F) nas áreas de monitoramento.
A partir dos dados de sequenciamento de 16S das amostras de solo coletadas nas diferentes áreas-fonte da Fazenda Ressacada, procedeu-se à realização de análise da diversidade funcional existente em solo, buscando genes envolvidos nos processos de biodegradação de hidrocarbonetos aromáticos e, também, associados ao metabolismo de enxofre e detoxificação. No que tange aos genes associados ao consumo de hidrocarbonetos, foram enfocados genes relacionados ao consumo de tolueno, quanto de intermediários de degradação de aromáticos, tanto por via aeróbica (ex. catecol), quanto anaeróbica (benzoato e benzoil-coA), conforme justificado a seguir.
Em bactérias aeróbicas, o oxigênio não atua apenas em um receptor terminal de elétrons para a conservação da energia respiratória, mas também se constitui em um reagente indispensável no mecanismo de ativação, mecanismo esse mediado pelas enzimas di-oxigenases e que envolve a formação de metabólitos intermediários centrais da via de degradação dos hidrocarbonetos, tais como o catecol (WIDDEL; RABUS, 2001). Posteriormente, entram em ação novamente as enzimas di-oxigenases para simplificar a molécula, com subsequente degradação via ciclo dos ácidos tricarboxílicos (EL-NAAS; ALHAIJA; AL-ZUHAIR, 2014).
Já as condições anaeróbicas – propícias à ferro, sulfato-redução e metanogênese – fornecem pouca energia às bactérias e geralmente não suportam a biodegradação completa dos compostos aromáticos, entretanto, o catabolismo anaeróbico pode atuar nos caminhos periféricos para convergir a uma via central, formando um intermediário aromático mais suscetível à degradação, normalmente benzoil-CoA (FOGHT, 2008).
A seguir é mostrada a porcentagem média de genes envolvidos em processos metabólicos, perante outros grupos gênicos envolvidos em outros processos encontrados nas 11 áreas experimentais:
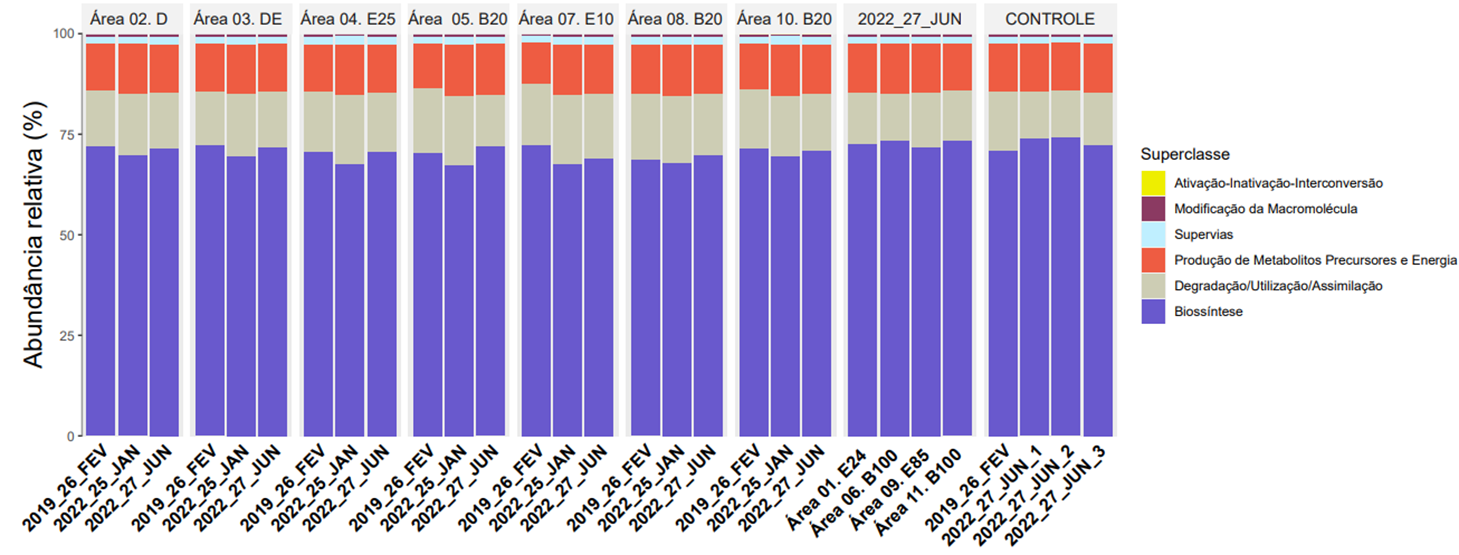
Abundância relativa dos genes da comunidade bacteriana envolvidos em cada um dos grandes grupos funcionais no solo próximo à fonte (F) nas áreas de monitoramento.
Na figura abaixo é possível visualizar a abundância de genes associados às principais vias de degradação de hidrocarbonetos associados às diferentes áreas. Percebe-se uma maior presença de genes associados à degradação de Benzoato nas Áreas 3 (DE_ANM), 5 (B20_ANM) e 10 (B20_BAF) de junho de 2022, enquanto as áreas 1 (E24_ANM), 6 (B100_ANM), 11 (B100_BFM) e Controle 3 não apresentaram a presença de genes para essa finalidade. As demais áreas apresentaram valores intermediários de abundância gênica associada à degradação de Benzoato. A via de degradação Benzoil-CoA somente foi associada às Áreas 2, 4, 5, 7 e 8 (em janeiro de 2022) e para as Áreas 2, 8 e 10 (em junho de 2022), sendo mais significativa na Área 4 (em janeiro de 2022). As vias de degradação de catecol, tolueno e do metabolismo do enxofre estavam presentes em todas as amostras, sendo que as vias associadas à biodegradação do catecol e de tolueno predominaram, respectivamente, nas Áreas de 2 e 8 coletadas em junho de 2022. Para o metabolismo do enxofre, as Áreas 4 e 5 apresentaram valores maiores nos dois períodos de avaliação em 2022. Outras amostras também apresentaram valores médios a altos, incluindo o Controle 1. Para a via de destoxificação, foram as amostras de solo das Áreas 2, 4, 7, 9 e 10 coletadas em junho de 2022 que apresentaram os maiores valores, enquanto nas amostras das Áreas 3 e 6 o potencial de ocorrência de tal via não foi observado.
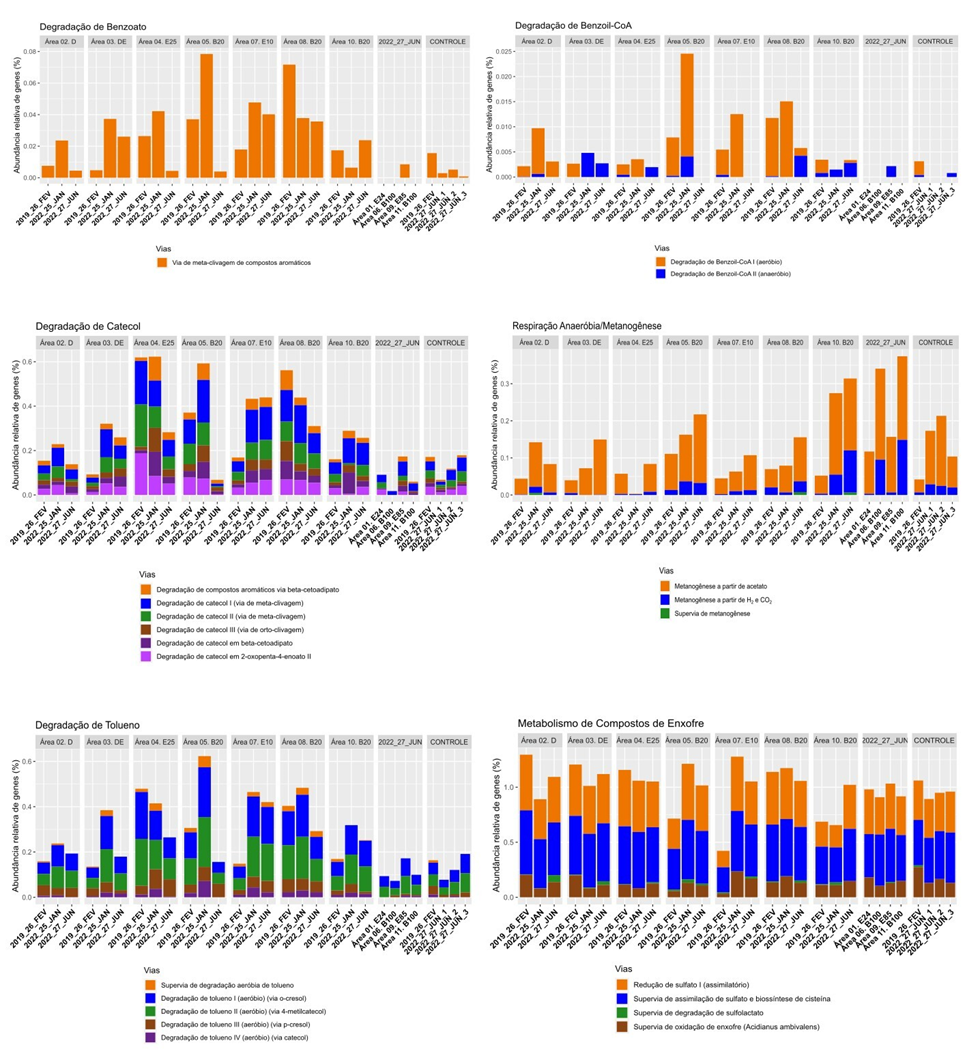
Abundância de genes da comunidade bacteriana envolvidos em processos metabólicos associados à degradação de hidrocarbonetos no solo próximo a fonte (F) nas áreas de monitoramento.
Para a via de degradação do catecol (Catechol Degradation), que agrupa uma série de vias metabólicas responsáveis pela degradação de hidrocarbonetos aromáticos, foi verificada a participação de vários grupos bacterianos. A família com maior participação foi Micrococcaceae com 28,7%, seguido de Micronosporaceae (19,7%), Oxalobacteraceae (17,4%) e Paenibacillaceae (14,1%). Além desses citados acima, Beijerinckiaceae, Comamonadaceae e Sphingomonaceae também tiveram uma participação nos processos metabólicos associados às vias metabólicas de degradação de hidrocarbonetos. Já para a via de degradação de benzoil-CoA (Benzoyl-CoA Degradation), somente a família Geobacteraceae apresentou integrantes potencialmente ativos. Os membros da família Micrococcaceae, como Arthrobacter phenanthrneivorans (KALLIMANIS et al., 2009) e Arthrobacter oxydans (THION et al., 2013), têm sido associados à degradação de HPA, tanto em cultura pura quanto no ambiente (ARYAL; LIAKOPOULOU-KYRIAKIDES, 2013). Os gêneros da família Oxalobacteraceae podem ser encontrados de forma onipresente em amostras de solo e água e são conhecidos por sua capacidade de degradar hidrocarbonetos aromáticos de forma aeróbia (LEE; LEE, 2001; PÉREZ-PANTOJA et al., 2012) e anaeróbia (KIM et al., 2014), além da sua capacidade de reduzir o nitrato em condições anaeróbicas (MULLER et al., 2006; KIM et al., 2014). Outras bactérias como Pseudomonas, Sphingomonas e Mycobacterium spp. também são muito comuns em ambientes contaminados, pois atuam em diversos processos de degradação de hidrocarbonetos de petróleo (BASTIAENS et al., 2000; JOHNSEN; KARLSON, 2005). Os integrantes da família Geobacteraceae pertencem ao Filo Proteobacteria, Ordem Desulfuromonadales (LOVLEY et al., 2011). Os integrantes dessa família são mesófilos quimiorganotróficos anaeróbicos, não fermentadores, que possuem como característica marcante a capacidade de reduzir Fe(III) e Mn(IV) insolúveis. Esses organismos geralmente dominam em ambientes de redução de ferro, em particular em ambientes sujeitos a influências antropogênicas. As características fisiológicas das Geobacterespécies são empregadas em biotecnologia ambiental, como biorremediação de hidrocarbonetos aromáticos, metais pesados e organohalóides, bem como na geração de bioenergia em células de combustível microbianas e células de eletrólise microbiana (MAROZAVA et al., 2014). Os resultados dos monitoramentos das diversidades taxonômica e funcional obtidos nas 11 áreas experimentais ajudaram não apenas na avaliação das comunidades bacterianas e potencialidades em termos de biodegradação, como auxiliaram, juntamente com os dados químicos, a tomada de decisão quanto às intervenções necessárias em cada caso. As famílias bacterianas atuantes nos processos de degradação de hidrocarbonetos nas áreas de estudo estão listadas na figura a seguir.
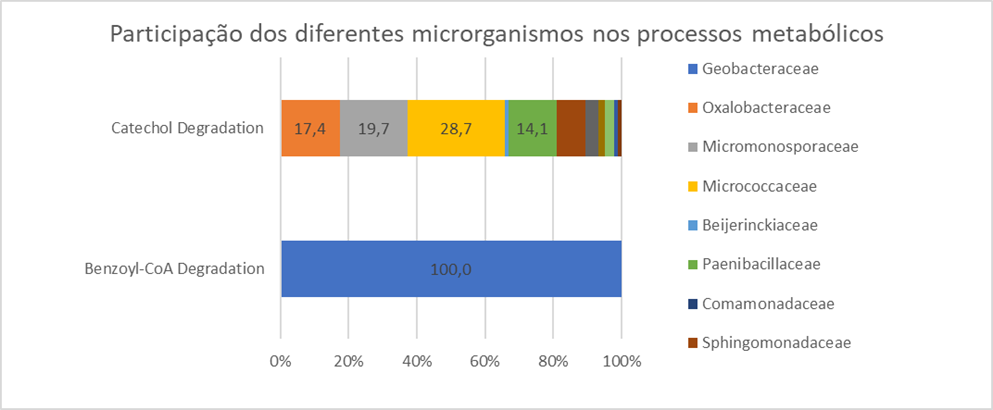
Participação das diferentes famílias bacterianas envolvidos nas vias de degradação do catecol (Catechol Degradation) e de benzoil-coenzimaA (Benzoyl-CoA Degradation).
3.2 Sequenciamento metagenômico da região ITS
Quanto aos fungos, uma alta porcentagem de organismos isolados é capaz de crescer em misturas complexas de hidrocarbonetos como única fonte de carbono, podendo ser utilizados como estratégia para melhorar ambientes poluídos (SINGH, 2006). O sequenciamento da região ITS permite a identificação de organismos com elevada resolução, associando a comunidade fúngica ao ambiente estudado. Nas áreas experimentais avaliadas, foram encontrados 12 filos, 39 classes, 75 ordens, 145 famílias, 214 gêneros e 426 ASV fúngicos com diversas funcionalidades.
Para os fungos, em nível de Família foram encontradas como predominantes Pseudeurotiaceae na Área 8 e 10, contaminadas com misturas B20 no monitoramento de fevereiro de 2019. Já na Área 5, houve predomínio de Giomerellaceae. A abundância relativa ultrapassou os 50% de Microascaceae na Área 8 em janeiro de 2022 e na Área 10 em junho de 2022. Por sua vez, o predomínio de Thichosphaeriaceae foi evidente na Área 6 (com 100% de biodiesel), sob processo de atenuação natural. A abundância relativa ultrapassou os 75% de Hypocreaceae nas Áreas 2, 3 e 5 no monitoramento de junho de 2022.
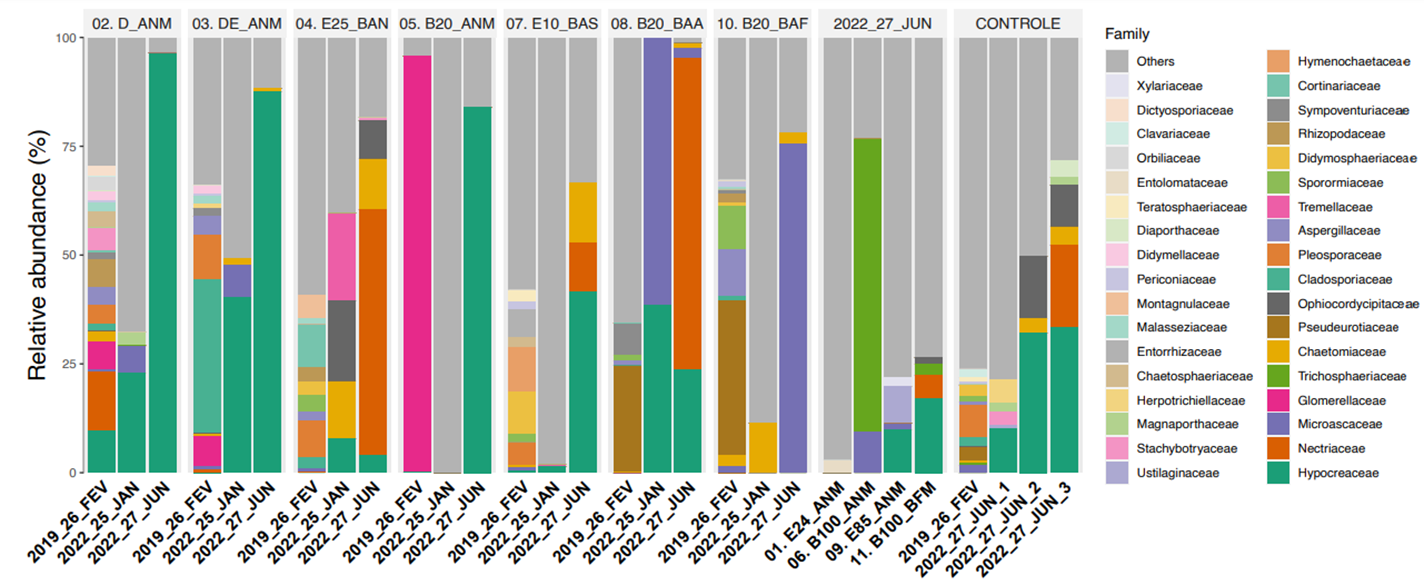
Abundância relativa de fungos em nível de Família no solo próximo a fonte (F) nas áreas de monitoramento.